This material is based upon work supported by the US National Science Foundation under Grant Number CHE-0848397. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
 Visual Molecular Dynamics (VMD) Software
Visual Molecular Dynamics (VMD) Software
Visual Molecular Dynamics (VMD) is a powerful, freely available software for the modeling, visualization and analysis of biomolecules available across multiple platforms. VMD allows researchers to quickly and easily represent structures in a fully three-dimensional space and is compatible with Protein Data Bank (PDB) and CCDC files. The VMD software can be downloaded from the Theoretical and Computational Biophysics Group at the University of Illinois at Urbana-Champaign.
Below are examples of biomolecules that you can view using VMD. Anthony Giovengo put a lot of effort into preparing these files together with Dr Brasch, and deserves a special thank you for this work! The images on this page are just 2D previews. Using VMD, you will be able to freely rotate the molecules in space to look at them from different angles on your computer screen. This will allow a better understanding of the molecules’ structures than any two-dimensional representation could give.
First you have to download and install the software, as well as download the files for the molecules you want to view. When starting VMD, it immediately opens three windows: a main window, a display window, and a status window you probably won’t need. But don’t close any of the windows while working with VMD! Use the main window to open a PDB file (File/New Molecule, then browse for the file name and click Load). Since VMD has opened a fourth window, you need to go back to the main window and open a VMD file corresponding to the PDB file you have opened (File, Load Visualization State, then browse for the file name). Now go to the display window, and you should see your molecule, which you can rotate by holding the left mouse button down, and moving the mouse.
 Aquacobalamin
Aquacobalamin
Aquacobalamin is a cobalamin (Cbl) derivative in which a water ligand is coordinated to the β-axial site of the complex (X = H2O, Figure 1). Aquacobalamin is one of the major forms of vitamin B12 isolated from mammalian cells.
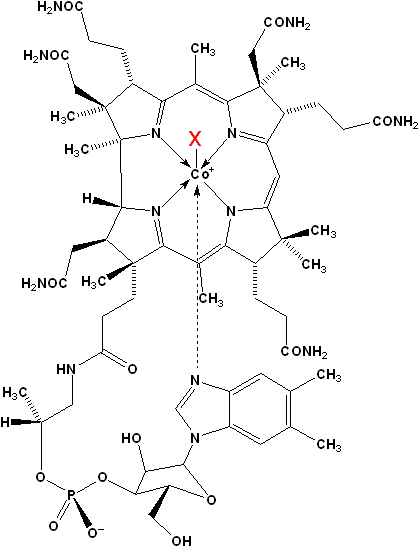
Figure 1: Structures of Cobalamins found in Humans:
X = H2O aquacobalamin (H2OCbl+)
X = CH3 methylcobalamin (MeCbl)
X = 5′-deoxyadenosine adenosylcobalamin (AdoCbl, “coenzyme B12“)
X = SO3 sulphitocobalamin (SO3Cbl)
X = CN cyanocobalamin (CNCbl, “vitamin B12“)
In the representation you can download, the cobalt atom is presented in yellow while the nitrogen atoms are shown in dark blue. The carbon and oxygen atoms are displayed in light blue and red, respectively. This structure is unpublished (Brasch group).
 MM-CoA mutase, Representation 1
MM-CoA mutase, Representation 1
 MM-CoA mutase, Representation 2
MM-CoA mutase, Representation 2
L-Methylmalonyl-coenzyme A mutase (MM-CoA mutase) is a B12-dependent enzyme which requires the B12 cofactor adenosylcobalamin (X = 5’-deoxyadenosyl, Figure 1). MM-CoA mutase catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA, an important intermediate in the TCA cycle.
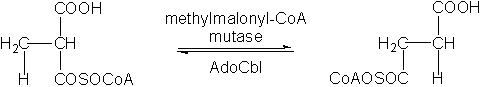
MM-CoA mutase from Propionibacterium shermanii is a α,β heterodimer (MM-CoA mutase in humans is an α2 dimer), with 1 Cbl cofactor and 1 substrate molecule (desulpho-CoA) bound on the α subunit (in humans 1 substrate and 1 Cbl cofactor are located on each subunit). The α subunit consists of 3 domains: a N-terminal upper substrate-binding domain comprised of a core of 8 twisted parallel β-strands surrounded by 8 α-helices called a (α,β)8 TIM barrel, a long linker chain and a C-terminal lower B12 binding domain, which is made up of 5 parallel β-strands and 6 α-helices in a flavodoxin-like fold (see below for a VMD image of flavodoxin).
In the first representation α-helices are shown in purple, β-sheets are in yellow, the desulpho-CoA substrate is lime green, and Cbl is displayed as a red stick diagram. A histidine from the protein displaces the 5,6-dimethylbenzimidazole coordinated to the α-axial site of the Cbl (= “base-off, His-on” Cbl) and is shown as a ball and stick model representation. For this histidine, nitrogen atoms are displayed in dark blue, carbon atoms in light blue and oxygen atoms are in red.
The second representation of MM-CoA mutase emphasizes the 4 subunits of the MMA-CoA mutase structure, highlighted in grey, gold, red and blue.
This structure was published in F. Mancia et al Structure 1996, 4, 339. The PDB code is 1REQ.
(Download PDB)
(Download VMD 1)
(Download VMD 2)
 Flavodoxin
Flavodoxin
Flavodoxins are electron transfer proteins that have a characteristic fold. The tertiary structure is comprised of a five-stranded parallel β sheet, displayed in yellow, surrounded by α helices at either side of the sheet, which are shown in purple. In this representation, the FMN cofactor is presented in an orange colored stick model.
This structure was published in C. L. Drennan et al J. Mol. Biol. 1999, 294, 711. The PDB code is 1CZN.
 Methionine synthase, Representation 1
Methionine synthase, Representation 1
 Methionine synthase, Representation 2
Methionine synthase, Representation 2
Methionine synthase is a mammalian B12-dependent enzyme which requires methylcobalamin (X = CH3, Figure 1) as a cofactor. In this reaction the methylation of homocysteine (Hcy) by 5-methyltetrahydrofolate to generate methionine and tetrahydrofolate is catalyzed by MeCbl-dependent methionine synthase.
Elevated levels of homocysteine have been linked to several diseases, including cardiovascular and neurodegenerative diseases, such as Alzheimer’s disease.
Methionine synthase (MS) from Escherichia coli is an α2 dimer. The upper domain in each subunit consists of an N-terminal 4-helix domain which functions as a cap in that it can block access to β side of Cbl in the resting state. Essentially, the upper domain must be displaced when the active site is presented to Hcy, CH3H4folate or S-adenosylmethionine (SAM is involved in reactivation of oxidized MS).The lower domain, the Cbl-binding domain, is comprised of 5 parallel β-strands and 6 α-helices with a flavodoxin-like fold structure. Once again a histidine residue from the enzyme displaces the 5,6-dimethylbenzimidazole at the α-axial site (“base-off, Cbl-on”). The lower side of the corrin ring is in contact with 3 loops of the enzyme. One of these loops provides the histidine while another one contains a conserved Leu which interacts with a specific CH3 group of the corrin ring.
In the first representation α-helices are shown in purple, β-sheets are in yellow, Cbl is displayed as an orange stick diagram and a detailed ball and stick model representation of the histidine is shown. In this histidine, nitrogen atoms are displayed in dark blue, carbon atoms in light blue and oxygen atoms are in red.
In the second representation, the emphasis is placed on the 2 subunits highlighted in red and blue respectively, with the remaining structure unaltered from the other representation.
This structure was published in C. L. Drennan et al Science 1994, 266, 1669. The PDB code is 1BMT.
(Download PDB)
(Download VMD 1)
(Download VMD 2)
 Holo-transcobalamin
Holo-transcobalamin
Holo-transcobalamin is a B12-binding protein transcobalamin (TC, TCII) that serves a vital role in cobalamin transport in plasma and intracellular uptake. Recombinant human TC is a homodimer with a cobalamin bound in each subunit. Each TC unit can be divided into two domains: an N-terminal α-domain and the C-terminal β-domain, linked by a short flexible linker. The α-domain is approximately 300 residues long and is almost entirely helical in nature, with 12 α-helixes shown in white and a 3/10 helix shown in blue. The His-172 and His-173 residues of the α-domain, represented as red stick diagrams, are the two most likely candidates for cobalt coordination at the β-axial site. Note that aquacobalamin was used in the preparation of holo-TC, and that the β-axial water ligand of aquacobalamin is readily displaced by stronger nucleophiles such as the imidazole of histidine from the protein. The β-domain spans approximately 100 residues a 5-stranded β-sheet, shown in yellow, packed against a 3-stranded β-sheet and a single α-helix. The base-on cobalamin complexes located between the two domains are represented as stick diagrams with their cobalt center in brown. Water molecules are represented as red spheres.
This structure was published in J. Wuerges et al Proc. Natl. Acad. Sci. USA 2006, 103, 4386. The PDB code is 2BB5.
 Holo-Intrinsic Factor
Holo-Intrinsic Factor
Holo-Intrinsic Factor is a cobalamin-binding protein intrinsic factor (IF) that plays an essential role in the absorption of cobalamins from the small intestine. IF itself is secreted by parietal cells of the stomach. The crystal of human recombinant IF-Cbl complex in this representation contains four molecules in the asymmetric unit. Two of these molecules are almost full length (residues 7–399 with a bound Cbl), while the other two are truncated (residues 7–273 and no bound Cbl). The full-length molecule contains two domains: an α-domain containing ~270 residues (7–273) and a β-domain comprising ~110 residues (289–399). The Cbl-binding pocket is at the interface of these two domains in an area with poor solvent accessibility. The defining feature of the α-domain is an intertwined α6/α6 helical barrel comprised of an inner core of 6 parallel even-numbered α-helices surrounded by an outer shell of 6 parallel odd-numbered α-helices running in the opposite direction. A short 3/10 linker is located at the bottom of the barrel and three disulfide linkers cross-link the α-domain. The β-domain contains a 4 stranded antiparallel β-sheet in addition to an α-helix and 3 β-sheets stacked above. A glycosylation site is located at residue Asn-395. Each full length molecule has the capacity for cobalamin binding. Two molecules of N-acetylglucosamine, a monosaccharide derivative of glucose, are shown as a lime green stick diagrams. In this representation, water molecules are displayed as red spheres, and penta-coordinate cob(II)alamin is shown as an orange stick diagram. α-Helices and β-sheets are purple and yellow respectively.
This structure was published in Mathews, F. S. et al Proc. Natl. Acad. Sci. USA, 2007, 104, 17311. The PDB ID is 2PMV.
 Holo-Intrinsic Factor bound to Cubilin
Holo-Intrinsic Factor bound to Cubilin
Holo-Intrinsic Factor bound to Cubilin – The ileal cubam receptor comprises the proteins cubulin and the transmembrane protein amnionless. This multi-ligand surface receptor is involved in receptor-mediated endocytosis of IF-B12 complexes. Four domains of the 27-domain cubulin, CUB5-8, interact with IF-B12. Of these domains, CUB6 directly interacts with the α-domain of IF and CUB8 with the β-domain. The other two CUB domains provide structural support roles. The conformation of IF-B12 is very similar to free IF-B12 and B12 itself is not recognized directly by the receptor. In this representation N-acetylglucosamine, penta-coordinate cob(II)alamin, β-D-mannose and α-D-mannose are shown as stick diagrams in lime green, orange, light blue and peach, respectively. Water and calcium ions are shown as red and silver spheres and α-helices and β-sheets in purple and yellow.
This structure was published by C. B. Folsted Andersen et al Nature 2010, 464, 445. The PDB code is 3KQ4.
Metals and Oxidative Stress